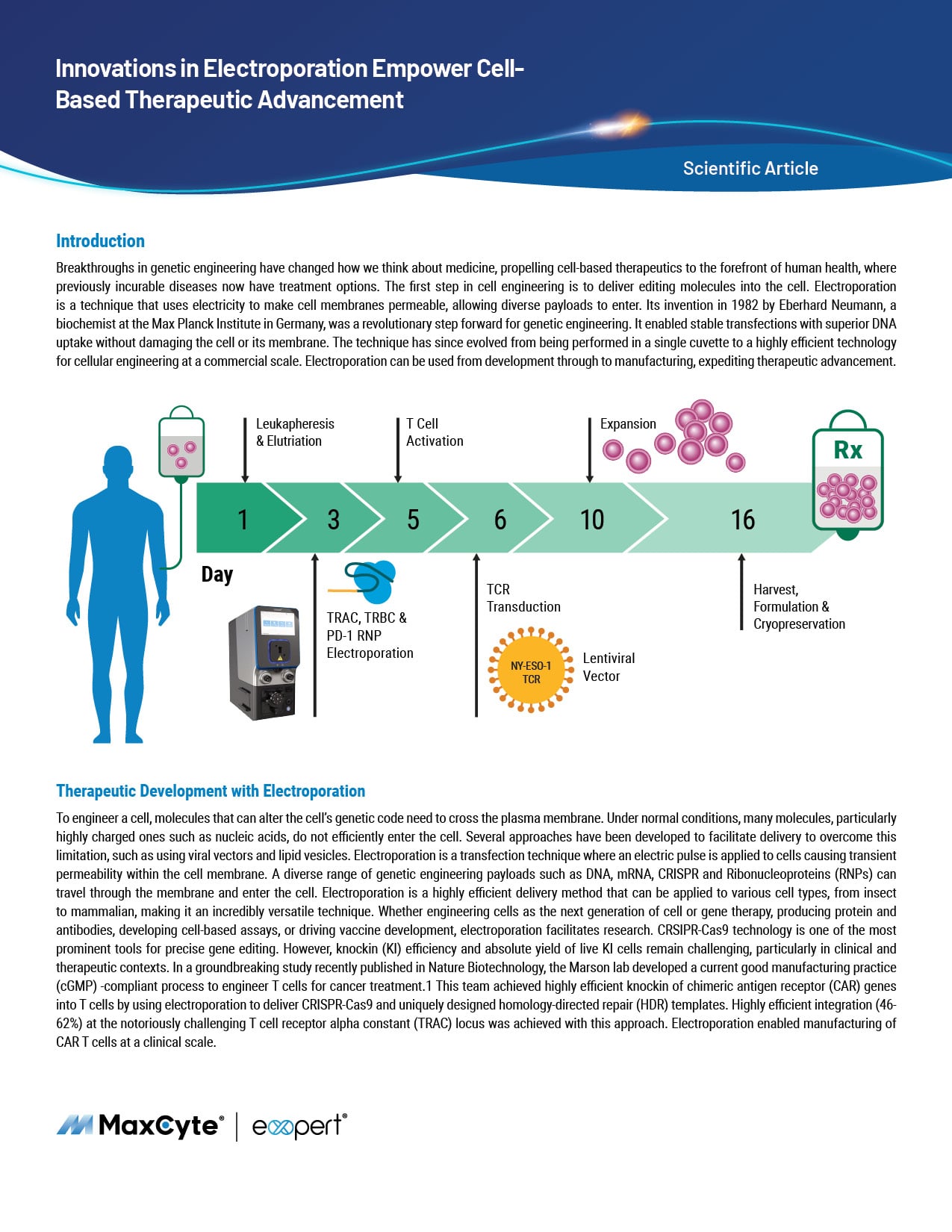
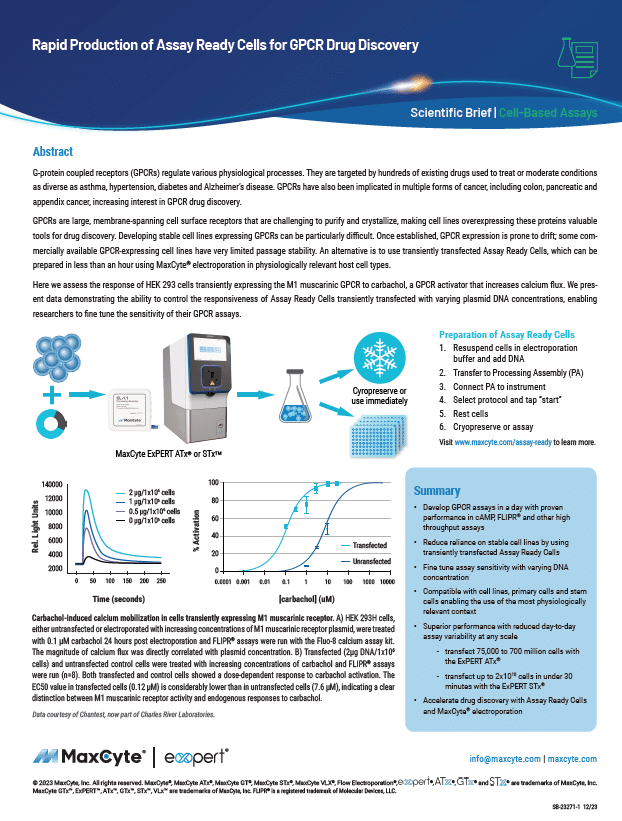
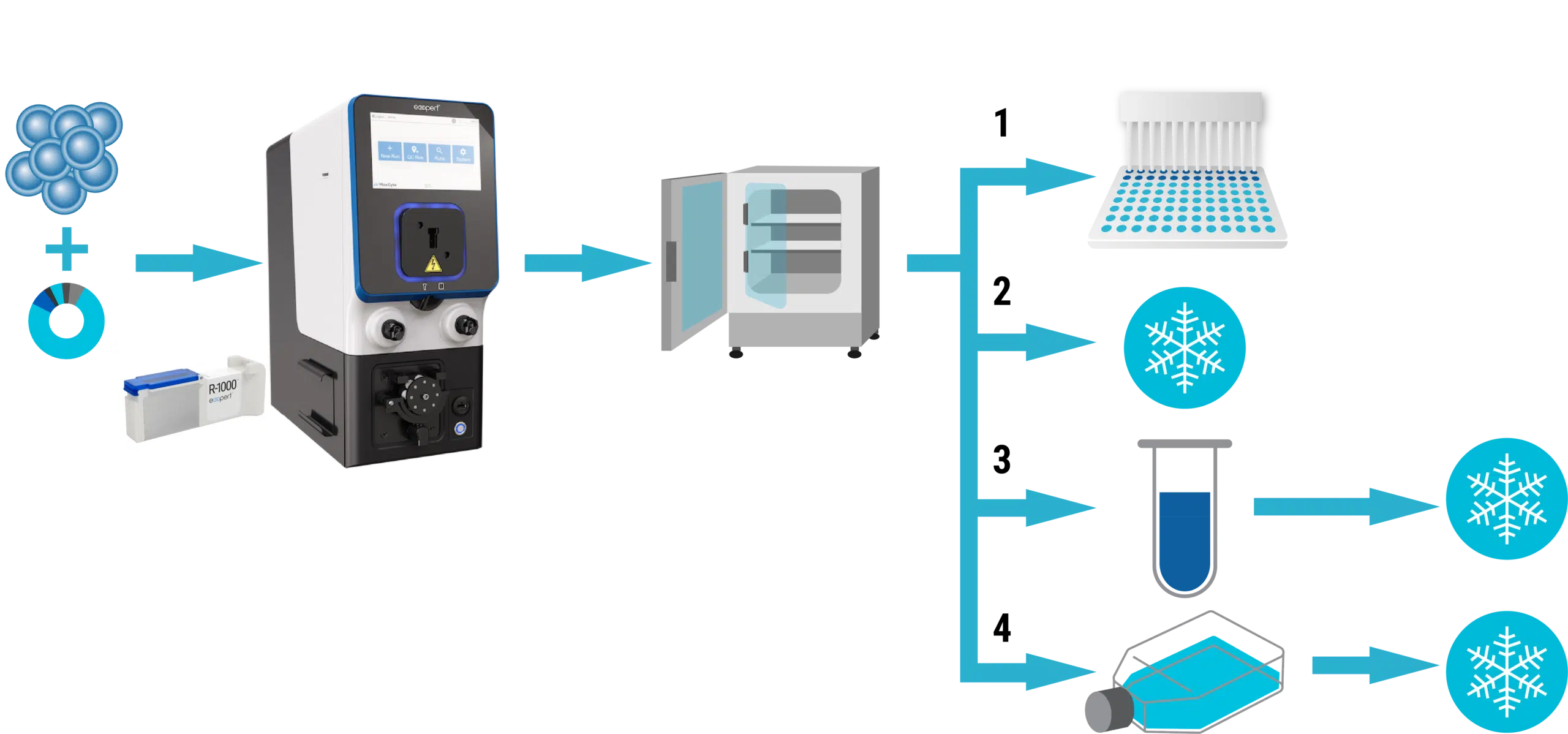
Resource Library
Technology alone won't get you where you want to go. And that's why we won't ever just offer technology.
Filter by
Resource type
- Application Note
- Article
- Brochure
- eBook
- Primary Literature
- Scientific Brief
- Scientific Poster
- Webinar/Presentation
Application
- Cell Based Assays
- Cell Line Development
- Cell Therapy
- CRISPR/Gene Editing
- Protein Production
- Stem Cells
- Vaccines
Cell type
- B Cells
- Cancer Cell Lines
- Cardiomyocytes
- CHO
- Dendritic Cells
- Fibroblasts
- HEK293
- HSPCs
- Insect Cell Lines (S2/Sf9/Sf21)
- ion channels
- iPSCs
- Keratinocytes
- MSCs
- Neurons
- NK Cells
- NPCs/NSCs
- Other Mammalian Cell Lines
- PBMCs
- T Cells